The contents of available preformatted databases are summarized separately according to their sequence nature and sources for nucleotide. The first section of videos were created by members of Dr.
2
Sura Yasin PDF Yasin Sharif Arabic English.

. And version 36 now includes lalign36 a program that finds multiple local alignments using Webb Millers sim algorithm and implementation of the Waterman and Eggert approach. FASTA and BLAST are the software tools used in bioinformatics. Wa law nashaaau lata masna alaaa aiyunihim fasta baqus-siraata fa-annaa yubsiroon.
It was no more than a single mighty Blast and behold. Ncbi blastチュートリアル このチュートリアルではncbiサイトでのblastによる相同性検索の方法について一般的な使い方を紹介してい ます はじめに. It is possible to use completely unstructured or even blank FASTA definition lines but this is not the recommended procedure.
The FASTA package is available from the University of Virginia and the European Bioinformatics Institute. Files UniGene SwissProt Files in the supported formats can be iterated over record by record or indexed and accessed via a Dictionary interface. Both BLAST and FASTA use a heuristic word method for fast pairwise sequence alignment.
It works by finding short stretches of identical or nearly identical letters in two sequences. The format originates from the FASTA software. The main application of SortMeRNA is filtering rRNA from metatranscriptomic data.
The makeblastdb application produces BLAST databases from FASTA files. Version 36 of the FASTA programs includes several major improvements. You can order a free printed copy from our colleagues at Cell Signaling Technology.
This note explains the following topics. Working of FASTA and BLAST. Statistical estimates are much more accurate.
Alas for My Servants. The file name extension gz is. Both from standalone and WWW Blast Clustalw FASTA GenBank PubMed and Medline ExPASy files like Enzyme and Prosite SCOP including dom.
Seqkit seq afasta bfasta seqkit seq --infile-list file-listtxt seqkit seq --infile-list. Threaded versions of the program are more efficient. SortMeRNA takes as input files of reads fasta fastq fastagz fastqgz and one or multiple rRNA database files and sorts apart aligned and rejected reads into two files.
What is bioinformatics Molecular biology primer Biological words Sequence assembly Sequence alignment Fast sequence alignment using FASTA and BLAST Genome rearrangements Motif finding Phylogenetic trees and Gene expression analysis. Recent advances in ultra-high-throughput sequencing technology and metagenomics have led to a paradigm shift in microbial genomics from few genome comparisons to large-scale pan-genome studies at. The FASTA file format used as input for this software is now largely used by other sequence database search tools such as BLAST and sequence alignment programs Clustal T-Coffee etc.
The db subdirectoryThis subdirectory contains a common set of preformatted BLAST database files in version 5 format. Below are links to online video lectures and tutorials for multiple versions of MEGA. MEGA is an integrated tool for conducting automatic and manual sequence alignment inferring phylogenetic trees mining web-based databases estimating rates of molecular evolution and testing evolutionary hypotheses.
They were like ashes quenched and silent. The classification annotation and sequences of human fly. Additional material and links.
Sudhir Kumars lab at the Institute for Genomics and Evolutionary Medicine at Temple UniversityThe rest of the videos were produced by users of MEGA. Assigning a unique identifier to every sequence in the database allows you to retrieve the sequence by identifier and allows you to associate every sequence with a. The FASTA sequences for a few widely used databases are stored under the FASTA subdirectory.
The output can be customized with the --format. The speed and sensitivity of the search can be adjusted with -s parameter and should be adapted based on your use case see setting sensitivity -s parameterA very fast search would use a sensitivity of -s 10 while a very sensitive search would use a sensitivity of up to -s 70A detailed guide how to speed up searches is here. These short strings of characters are called words.
The format also allows for sequence names and comments to precede the sequences. In bioinformatics and biochemistry the FASTA format is a text-based format for representing either nucleotide sequences or amino acid protein sequences in which nucleotides or amino acids are represented using single-letter codes. Sciences STKE has commentary and related articles in a special kinome issue.
A Human Kinome poster 19 Mb accompanies the article featuring a dendrogram of human protein kinases see picture above. Introduction to Bioinformatics Lecture.
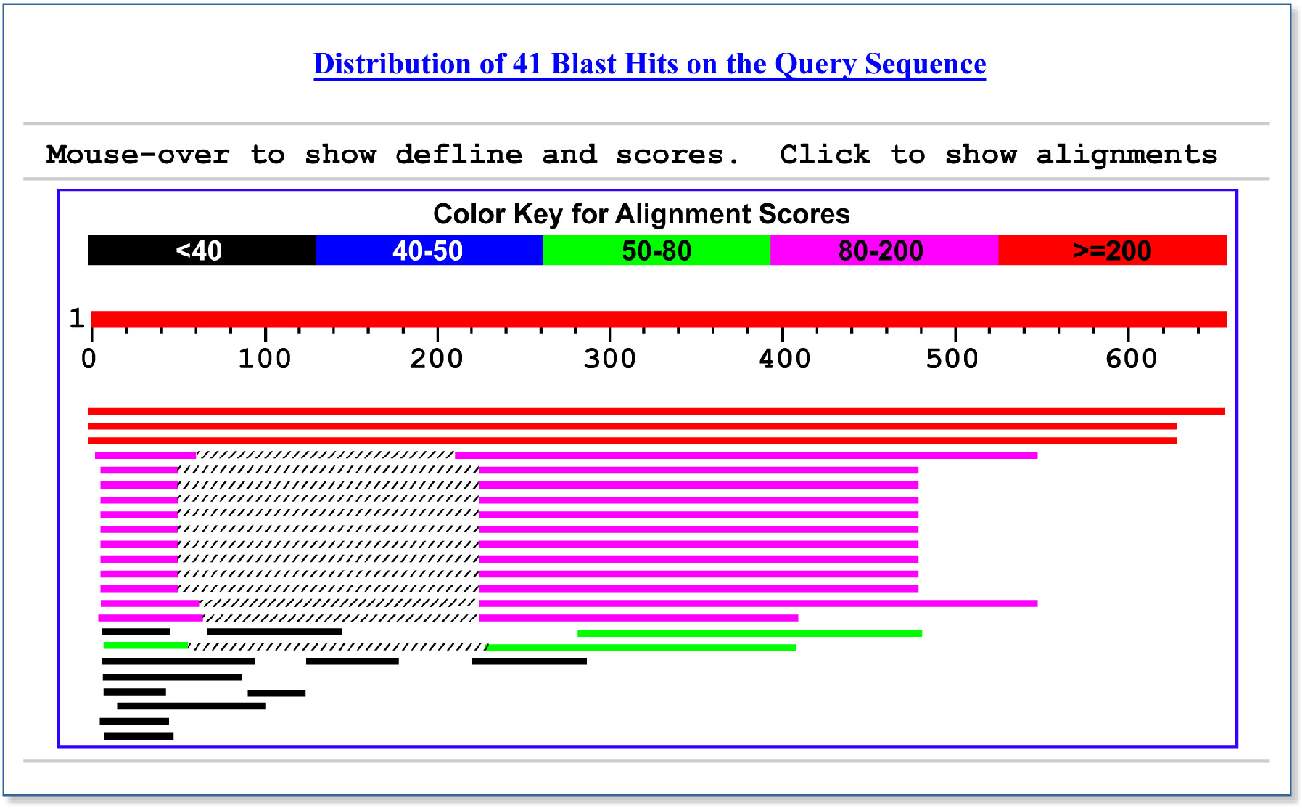
Pdf Tom Madden The Blast Sequence Analysis Tool 16 1 16 The Blast Sequence Analysis Tool Semantic Scholar
Pdf Tom Madden The Blast Sequence Analysis Tool 16 1 16 The Blast Sequence Analysis Tool Semantic Scholar

Pdf Tom Madden The Blast Sequence Analysis Tool 16 1 16 The Blast Sequence Analysis Tool Semantic Scholar
2

0 Comments